Resources
MacCoss Lab Mass Spectrometry Instrumentation
The MacCoss Lab maintains a state-of-the-art mass spectrometry facility equipped with cutting-edge instrumentation for both targeted and untargeted quantitative proteomics. Our instruments support a wide range of acquisition modes including targeted selected reaction monitoring (SRM), parallel reaction monitoring (PRM), data-independent acquisition (DIA), and data-dependent acquisition (DDA).
Capabilities
Our instrumentation suite enables:
- Targeted Proteomics: SRM and PRM workflows for precise and accurate quantification
- Untargeted Proteomics: DIA and DDA methods for discovery proteomics
- Clinical Applications: Robust, reproducible assays for clinical research experiments
- Method Development: Optimization of acquisition parameters and workflows
Thermo Fisher Orbitrap Astral
State-of-the-art mass spectrometer optimized for untargeted data independent acquisition-mass spectrometry

Example publications from our lab describing the use of the Orbitrap Astral and Astral Zoom for quantitative proteomics.
- Hsu et al., Evaluation of a prototype Orbitrap Astral Zoom mass spectrometer for quantitative proteomics - Beyond identification lists bioRxiv 2025
- Heil et al, Evaluating the Performance of the Astral Mass Analyzer for Quantitative Proteomics Using Data-Independent Acquisition J. Proteome Res. 2023
Thermo Fisher Stellar Mass Spectrometer
Hybrid Quadrupole Mass Filter-Radial Ejection Linear Ion Trap

Example publications describing the Stellar from the MacCoss lab include:
- Plubell et al., Development of highly multiplex targeted proteomics assays in biofluids using the Stellar mass spectrometer bioRxiv 2024
- Remes et al., Hybrid Quadrupole Mass Filter - Radial Ejection Linear Ion Trap and Intelligent Data Acquisition Enable Highly Multiplex Targeted Proteomics J. Proteome Res. 2024
Thermo Fisher Orbitrap Eclipse Tribrid
Advanced tribrid mass spectrometer combining quadrupole, ion trap, and Orbitrap technologies

Thermo Fisher Orbitrap Exploris 480
High-performance Orbitrap mass spectrometer for comprehensive proteomics workflows

Thermo Fisher Orbitrap Fusion Lumos Mass Spectrometers x2
We operate two Fusion Lumos tribrid mass spectrometers for advanced proteomics. These instruments combine quadrupole, ion trap, and Orbitrap technologies and are a workhorse for proteomics technology development.


Thermo Fisher Q Exactive HF
High-field Orbitrap mass spectrometer for quantitative proteomics

Thermo Fisher TSQ Altis
High-performance triple quadrupole mass spectrometer for targeted quantitative analysis

MacCoss Lab Software Tools
Windows Client Tool for the visualization, analysis, and development of methods for quantitative mass spectrometry
- Free, open-source quantitative mass spectrometry software. Skyline is a freely-available, open-source Windows client application for building Selected Reaction Monitoring (SRM) / Multiple Reaction Monitoring (MRM), Parallel Reaction Monitoring (PRM), Data Independent Acquisition (DIA/SWATH) and DDA with MS1 quantitative methods and analyzing the resulting mass spectrometer data. Its flexible configuration supports All Molecules. Skyline uses state-of-the-art technologies for creating and iteratively refining targeted methods for large-scale quantitative mass spectrometry studies in life sciences.
- Supports proteomics, metabolomics, and small molecule workflows.
- Vendor agnostic. Can analyze data and generate methods for all major instrument vendors. Including Agilent, Bruker, Shimadzu, ThermoFisher, and Waters.
- Download at: skyline.ms
- External Tools: Skyline has an external tool framework. We have a tool store with 20 tools currently available.
- Source Code is available as part of the Proteowizard project.
- Original Publication MacLean et al, Skyline: an open source document editor for creating and analyzing targeted proteomics experiments, Bioinformatics 2010
- Cited: 5,315 times
Widely used software tools and libraries for mass spectrometry data analysis.
- ProteoWizard provides a set of open-source, cross-platform software libraries and tools (e.g. msconvert, Skyline, IDPicker, SeeMS) that facilitate proteomics data analysis. The libraries enable rapid tool creation by providing a robust, pluggable development framework that simplifies and unifies data file access, and performs standard chemistry and LCMS dataset computations.
- Download and Documentation: https://proteowizard.sourceforge.io/
- Source Code: Available on Github and licensed under Apache 2.0.
- Original Publication: Chambers et al, A cross-platform toolkit for mass spectrometry and proteomics, Nature Biotechnology 2012
- Cited: 4,490 times
Web-based repository for Skyline documents and colaboration
- Panorama is a freely-available, open-source webserver for sharing experiments and validated assays that integrates into a Skyline proteomics workflow. Panorama can be installed on a local server, or you can request a project on the PanoramaWeb.org server, hosted by the MacCoss Lab at the University of Washington. Access privileges within a project may be customized, allowing you to control fully who has access to data you publish to Panorama.
- Access: panoramaweb.org
- Panorama Public: One of six of the ProteomeXchange servers used by the proteomics community. Panorama Public simplifies the process of sharing datasets analyzed by Skyline.
- Requirements: Can be accessed within Skyline and from any modern browser (Chrome, Firefox, Safari, Edge)
- Features: Unique tools for data sharing, collaboration, quality control
- API: Programmatic access for automated workflows
Open Source Server for the Analysis and Sharing of Data Dependent Acquisition Mass Spectrometry Results
- Limelight is designed to provide you with the full-stack of proteomics results, regardless of which processing pipeline you used to search your data. Full-stack means that you have access to the global views of your data (such as statistically comparing conditions), to viewing lists of proteins and peptides, to individual PSMs and spectra–all showing the native scores from whichever pipeline you used. Additionally, all native scores from your pipeline are available to you for filtering–even when contrasting multiple searches that each used different pipelines.
- Limelight can be installed locally or you can request an account on a server hosted at the University of Washington.
- Detailed documentation for using Limelight is available here.
- Source Code is available on GitHub.
Open source tool for peptide-centric analysis of data independent acquisition-mass spectrometry data
- EncyclopeDIA is library search engine comprised of several algorithms for DIA data analysis and can search for peptides using either DDA-based spectrum libraries or DIA-based chromatogram libraries. Check out our manuscript describing EncyclopeDIA at Nature Communications (Searle et al, 2018) for more information. EncyclopeDIA contains Walnut, an implementation of the PECAN (Ting et al, 2017) scoring system, to enable chromatogram library generation from FASTA protein sequence databases when spectrum libraries are unavailable. EncyclopeDIA also supports Prosit, a deep learning tool for generating peptide fragmentation spectra, as described in (Searle et al, 2020). EncyclopeDIA also contains Thesaurus for localizing and quantifying PTMS with DIA experiments (Searle et al, 2019)
- Suport – EncyclopeDIA is maintained and supported by the Searle lab at the Mayo Clinic.
- Downloads and Documentation – Details can be found on the EncyclopeDIA Bitbucket page.
- Source Code is also available on the Bitbucket page under an Apache 2.0 license.

Comet is an open source fork of the original SEQUEST database tool for proteomics
- Searching uninterpreted tandem mass spectra of peptides against sequence databases is the most common method used to identify peptides and proteins. Since this method was first developed in 1993, many commercial, free, and open source tools have been created over the years that accomplish this task. Although its history goes back two decades, the Comet search engine was first made publicly available in August 2012 on SourceForge under the Apache License, version 2.0. The repository was migrated to GitHub in September 2021. Comet is maintained by Jimmy Eng at the UW Proteomics Resource.
- Download and Documentation are available on the UW Proteomics Resource Github.
- Support is available via a Google Groups.
- Source Code is available on GitHub under an Apache 2.0 license
The Crux mass spectrometry analysis toolkit is an open source project maintained by the Noble lab that aims to provide users with a cross-platform suite of analysis tools for interpreting peptide MS/MS data.
- The toolkit includes several search engines for both standard and cross-linked database search, as well as a variety of pre- and post-processing engines for assigning high-resolution precursor masses to spectra, assigning statistical confidence estimates to spectra, peptides and proteins, and performing label free quantification. Crux comes pre-complied for the Linux, Windows and MacOS operating systems. It is implemented as a single program that offers a wide variety of commands.
- Support is available via a Google Groups.
- Download and Documentation are available on the Crux website.
- Source Code is available on GitHub under an Apache 2.0 license
 Percolator
Percolator
Percolator: semi-supervised learning for peptide identification from shotgun proteomics datasets
- Percolator has become the gold standard for post-processing and FDR control for bottom-up proteomics. Our software is incorporated as part of Proteome Discoverer, FragPipe, Mascot, Crux, and many more. Percolator is actively maintained by the Käll lab.
- Original Publication Lukas Käll et al, Semi-supervised learning for peptide indentification from shotgun proteomics datasets Nature Methods 2007
- Download and Documentation are available on the Percolator website. Percolator is also part of the Crux project.
- Source Code is available on GitHub under an Apache 2.0 license.
Public Datasets on Panorama Public
We have made available a number of mass spectrometry datasets on Panorama Public
Browse all MacCoss Lab datasets on Panorama Public →
Last updated: April 01, 2026 — 61 datasets available
2026
2025
2024
2023
2022
2021
2020
2019
2018
2017
2015
All datasets include processed results as Skyline documents and raw datafiles. Many datasets are paired with published manuscripts.
Educational Materials
Last updated: April 01, 2026 — 28 Skyline tutorials available
UWPR Mass Spectrometry Resources
- UWPR LC-MS Tips and Tricks — Protocols, tips, and resources for LC-MS analyses. Definitely bookmark this page.
- UWPR Data Analysis Tools — Online calculators, database search tools, and computational resources.
UWPR Online Calculators
- MS/MS fragmentation calculator
- Protein sequence digestion calculator
- Isotope distribution calculator
- Elemental mass calculator
- Lorikeet Spectrum viewer
- Koina (Prosit, ms2pip, AlphaPeptDeep) spectrum prediction viewer
- Table of relevant masses (amino acids, elements)
Skyline Tutorials
Hands-on tutorials with real data and step-by-step instructions
Introductory
- Targeted Method Editing (26 pages)
- Targeted Method Refinement (28 pages)
- Grouped Study Data Processing (70 pages)
- Existing & Quantitative Experiments (43 pages)
Introduction to Full-Scan Acquisition Data
- Comparing PRM, DIA, and DDA (38 pages)
- PRM With an Orbitrap (44 pages)
- Basic Data Independent Acquisition (40 pages)
Full-Scan Acquisition Data
- MS1 Full-Scan Filtering (41 pages)
- DDA Search for MS1 Filtering (19 pages)
- Parallel Reaction Monitoring (PRM) (40 pages)
- Analysis of DIA/SWATH Data (32 pages)
- Analysis of diaPASEF Data (36 pages)
- Library-Free DIA/SWATH (26 pages)
- Peak Boundary Imputation for DIA (16 pages)
Small Molecules
- Small Molecule Targets (10 pages)
- Small Molecule Method Development (37 pages)
- Small Mol. Multidimension Spec. Lib. (23 pages)
- Small Molecule Quantification (27 pages)
- Hi-Res Metabolomics (17 pages)
Reports
- Custom Reports (33 pages)
- Live Reports (48 pages)
Advanced Topics
- Absolute Quantification (19 pages)
- Advanced Peak Picking Models (28 pages)
- iRT Retention Time Prediction (36 pages)
- Collision Energy Optimization (12 pages)
- Ion Mobility Spectrum Filtering (26 pages)
- Spectral Library Explorer (22 pages)
- Audit Logging (23 pages)
Skyline Documentation
Advanced reference documentation and developer resources
- Skyline Custom Reports — Learn about the vast array of values you can show in the Document Grid or export
- Skyline Command-Line Interface — Use SkylineRunner.exe and SkylineCmd.exe for command-line operations
- Skyline Keyboard Shortcuts — Quick access to commands without leaving the keyboard
- External Tools Documentation — Integrate statistical and bioinformatics tools with Skyline
- Interactive Tools Documentation — Develop .NET tools that interact with Skyline in real-time
Skyline Videos
Quick instructional videos for getting started
- Video Demo 1: Creating SRM/MRM Methods (28 minutes)
- Video Demo 2: Results Analysis and Method Refinement (25 minutes)
- Video Demo 3: Importing Existing Experiments (27 minutes)
- Skyline Trailer Video
YouTube Channels
Course content and instructional videos
- Skyline Course at UW — University of Washington course materials
- May Institute at Northeastern University — Comprehensive proteomics course content
- Targeted Proteomics Course at ETH, Zurich — International course materials
LC-MS Tips and Protocols
Selected resources from the UWPR LC-MS Tips page
Support & Training
Last updated: April 01, 2026 — 4 upcoming events, 101 past events, 27 webinars
Forums and Discussion
- Skyline Support Board
- Panorama Support Board
- University of Washington Proteomics Listserv - If you are at UW and doing proteomics you should join this list.
Upcoming Events
- 2026 ISAS Dortmund Skyline Training Course (March 2-5, 2026)
- May Institute - Computation and statistics for mass spectrometry and proteomics
- Frontiers in Proteomics: Advanced Skyline Applications (July 6-10, 2026)
- Cascadia Proteomics Symposium (July 16-17, 2026)
Skyline Webinar Series
The Skyline Team presents tutorial webinars designed to help you get the most out of Skyline targeted proteomics software. Each ~90 minute webinar includes Q&A, presentations, and tutorial data.
Recent Webinars (2025):
- #27: Peak Boundary Imputation for DIA Data (Dec 2025)
- #26: DIA with FragPipe, DIA-NN and Skyline (Sept 2025)
- #25: Comparing Acquisition Methods (Jan 2025)
Past Events by Year
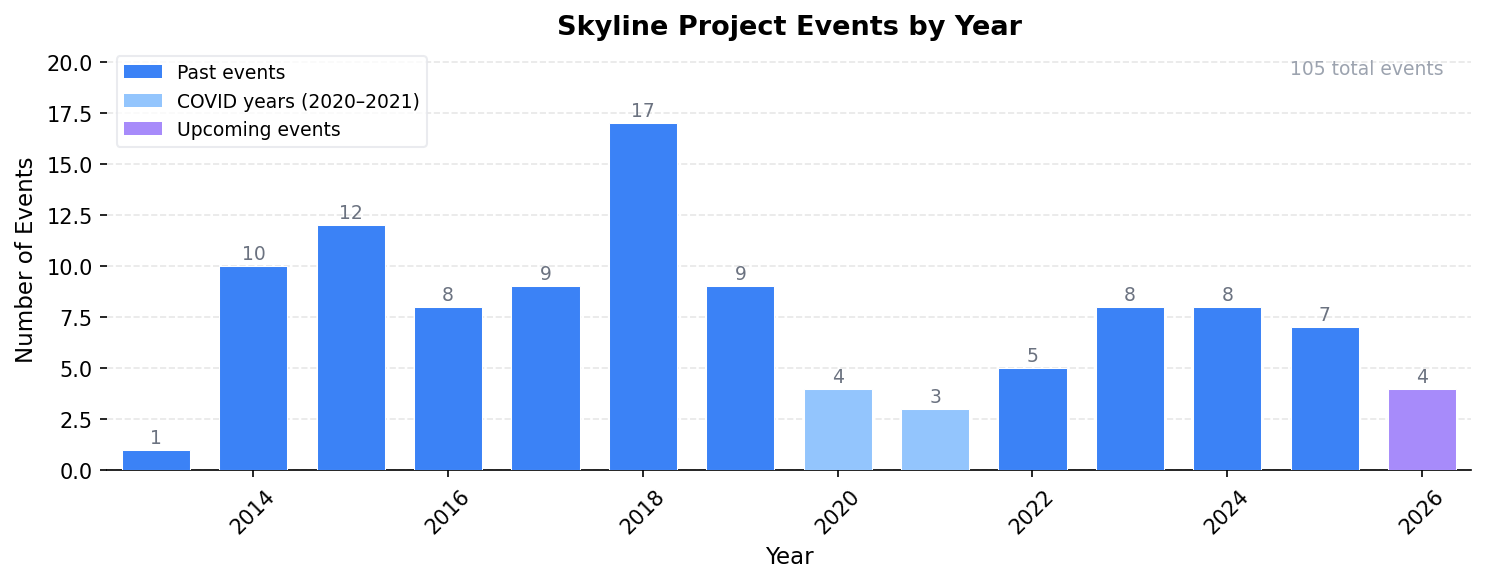
2025 Events
- Skyline Online 2025 (October 7-17, 2025)
- Practical Course on Targeted Proteomics - Barcelona, Spain (October 5-10, 2025)
- Skyline Course at the University of Washington - Seattle, WA (July 7 - 11, 2025)
- Two-day Short Course: 08 Quantitative Proteomics: Case Studies - Baltimore, MD
- Skyline User Group Meeting - Baltimore, MD (June 1, 2025)
- May Institute - Computation and statistics for mass spectrometry and proteomics - Northeastern University
- 2025 ISAS Dortmund Skyline Training Course - Dortmund (April 7-10, 2025)
Proteomics Services
The MacCoss Lab offers mass spectrometry-based proteomics services to researchers at the University of Washington and external collaborators. Our services leverage our state-of-the-art instrumentation and decades of expertise in quantitative proteomics.
Available Services
- Biofluid Proteomics: Analysis of plasma, serum, CSF, and other biofluids
- Targeted Assay Development: Custom SRM/MRM and PRM method development for specific proteins of interest
- Data-Independent Acquisition (DIA): Comprehensive proteome profiling using our latest instruments
- Method Consultation: Expert guidance on experimental design and sample preparation
Getting Started
For detailed information about our services, pricing, and to submit a project request, please visit our services portal:
Contact
For questions about our proteomics services, please contact us at services[at]maccosslab[dot]org